Functional Proteins from a Random-Sequence Library.
Abstract
Functional primordial proteins presumably originated from random sequences, but it is not known how frequently functional, or even folded, proteins occur in collections of random sequences. Here we have used in vitro selection of messenger RNA displayed proteins, in which each protein is covalently linked through its carboxy terminus to the 3′ end of its encoding mRNA1, to sample a large number of distinct random sequences. Starting from a library of 6 × 1012 proteins each containing 80 contiguous random amino acids, we selected functional proteins by enriching for those that bind to ATP. This selection yielded four new ATP-binding proteins that appear to be unrelated to each other or to anything found in the current databases of biological proteins. The frequency of occurrence of functional proteins in random-sequence libraries appears to be similar to that observed for equivalent RNA libraries2,3.
Acknowledgements
We thank members of the Szostak laboratory and especially D. Wilson, G. Cho, G. Short, J. Pollard, G. Zimmermann, R. Liu, J. Urbach and R. Larralde-Ridaura for their helpful advice. This work was supported in part by the NASA Astrobiology Institute and the NIH. J.W.S. is an investigator at the Howard Hughes Medical Institute.
Supplementary information
Generalized Random Library sequence (DNA)
TTCTAATACGACTCACTATAGGGACAATTACTATTTACAATTACAATGGACTACAAAGACGACGACGATAAGAAGACTYACTGZ(XYZ)18YACTGZ(XYZ)18YACTGZ(XYZ)18YACTGZ(XYZ)18YACTGGTCAGCGAGCTGCCATCATCATCATCATCATATGGGAATGTCTGGATCT
Average nucleotide composition of random parts of Random Library
- XYZ
A35330
T202922
G272149
C181729
(%)
Generalized Random Library sequence (Protein)
MDYKDDDDKKT(Random)81WSASCHHHHHHMGMSGS
Average amino acid composition of random part of Random Library (Protein)
- Ala 4.1 Leu 7.4
Arg 6.8 Lys 5.1
Asn 7.5 Met 4.5
Asp 5.5 Phe 2.8
Cys 4.6 Pro 2.8
Gln 2.6 Ser 6.6
Glu 4.0 Thr 5.4
Gly 5.3 Trp 4.4
His 3.8 Tyr 5.0
Ile 4.8 Val 7.1
(%)
Selected clones from round 8
Family A
- 08-05MNYKDDDDKKTHWYTNSGFAMTSLRFMMIKWYNWWHDQRHRNIRHHRAMAPRN
CRIQAITPTHGHDLPQSFEDWRRDYRYNRDKTMAKGYQPWSASCHHHHHHMGMSGS
08-07MDYKDDDDKKTHWYTNSGFAMTSLRFMMIKWYDWWHDQRHRNIRHHRAMAPRN
CRIQAITPTHGHDLPQSFEDWRWDYRYNRDKTMAKGYQPWSASCHHHHHHMGMSGS
08-09MDYKDDDDKKTHWYTNSGFAMTSLRFMMIKWHNWWHDQRHRNIRHHRAMAPRN
CRIQAITPAHGHDLPQSFEDWRWDYRYNRDKTMAKGYQPWSASCHHHHHHMGMSGS
08-48MDYKDDDDKKTHWYTNSGFAMTSLRFMMIKWYNWWHDQRHRNIRHHRAMAPRN
CRIQAITPTHGHDPPQSFEDWRWDYRYNRDKTMAKDYQPWSASCHHHHHHMGMSGS
Family B
- 08-01MDYKDDDDKKTNCHQKRIYRVKPCVICKVAPRDWWMENRHLRIYTMCKTCFSN
CINYGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
08-04MDYKDDDDKKTNWHQRRIYRVKPCVICKVAPRDWWVENRHLRIYTMCKTCFSN
CINSGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
08-08MDYKDDDDKKTNWHQKRIYRVKPCVICKVAPRDWWVENRHLRIYTMCKTCFSN
CINNGDDTYYGHDDWLMYTDCKEFSNTYHDLGRLPDEDRWSASCHHHHHHMGMSGS
08-10MDYKDDDDKKTNWHQKRIYRVKPCVICKVAPRDWWVENRHLRIYTMCKTCFSN
CINYGDDTYYGHDDWLMYTDCEEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
08-12MDYKDDDDKKTNCHQKRIYRVKPCVICKVAPRDWWVENGHLRIYTMCKTCFSN
CINYGDDTYYGHDDWLMYTDCKEFSNTYHNLDRLPDEDRWSASCHHHHHHMGMSGS
08-13MDYKDDDDKKTNWHQKRIYRVKPCVICKVAPRDWWVENRHLRIYTMCKTCFSN
CINYGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
08-14MDYKDDDDNKTNWHQKRIYRVKPCVICKVAPRDWWVENKHLRIYTMCKTCFSN
CINYGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
08-15MDYKDDDDKKTNWHQKRIYRVKPCVICKVAPRDWWVENRHLRIYTMCKTCFSN
CINYGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
08-18MDYKDDDDKKTNWHQERIYRVKPCVICKVAPRDWWVENRHLRIYTMCKTCFSN
CVNYGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
08-21MDYKDDDDKKTNWHQKRIYRVKPCVICKVAPRDWWVENRHLRIYTMCKTCFSN
CINNGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
08-23MDYKDDDDKKTNWHQKRIYRVKPCVICKVAPRDWWVENRRLRIYTMCKTCFSN
CINYGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
08-45MDYKDDDDKKTNWHQKRIYRMKPCVICKVAPRDWWVENRHLRIYTMCKTCFSN
CINYGDDTYYGHDDWLIYTDCKEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
08-46MDYKDDDDKKTNWHQKRIYRVKPCVICKVAPRDWWVENRHLRIYTMCKTCFSN
CINYGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
08-47MDYKDDDGKKTNWHQKRIYRVKPCVICKVAPRDWWVENRHLRIYTMCKTCFSN
CINYGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRWSASCHHHHHHMGMSGS
Family C
- 08-11MDYKDVDDKKTHCDKTVSVDMTFRVRNMKVAKDCWSVVVWTKRSNYFSGRQLH
CDSWHHYNSRRFGTETKLAYWELPKWKWKINNTHAINIHWSASCHHHHHHMGMSGS
08-17MDYKDIDDKKTHCDKAVSVDMTFRVRNMKVAKDCWSVVVWTKRSNYFNGRQLH
CDSWHHYNSRRFGTETKLAYWELPKWKWKINNTHAINIHWSASCHHHHHHMGMSGS
08-19MDYKDIDDKKTHCDKAVSIDMTFRVRNMKVAKDCWSVVVWTKRSNYFSGRQLH
SDSWHHYNSRRFGTETKLAYWELPKWKWKINNTHAINIHWSASCHHHHHHMGMSGS
08-06MDYKDVDDKKTHCDKAVSVDMTFRVRNMKVAKDCWSVVVWTKRSNYFSGRQLH
CDSWHHYNSRRFGTETKLAYWELPKWKWKINNTHAINIHWSASCHHHHHHMGMSGS
Family D
08-20MDYKDDDDKKTYWHALVTYNKTLSYRLATKFTDWWNLDPPRNMQTKVSELNLH
WLKSGGKGTQKAHSINEISNWVHQHELSDKSMRLHSKVRWSASCHHHHHHMGMSGS
Selected clones from round 18
18-01MDYKDDDDKKTNWQKRIYQVRPCVICKVAPRDWRVENRHLRIYNMCKTCFSNS
VDYGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-02MDYKDDDDKKTNWQKRVYRARPCVICKVAPRDWRVVNRHLRIYNMCKTCFSNS
INHGDDTYHGHNDWLMYTDCEEFSSTCHNLGRQPDEDRHWSASCHHHHHHMGMSGS
18-03MDYKDDDDKKTYWQKRIYRVRPCVICKVAPRDWRVKNGHLRIYNMCKTCFSNS
IKCGDDTYYGHDDWLIHTDCKDFSNTYLNLGRLPDEERHWSASCHHHHHHMGMSGS
18-04MDYKDDDDKKTNWQKRVYRVRPCVVCKEAPRDWRVKDRHLRIYNMCKTCFSNS
INYGDDTHYGHDDWLMYTDCKGFSNTYHNPSRLPDEDRHWSASCHHHHHHMGMSGS
18-05MDYKDDDDKKTNWQKRIYRVGPCVICKVAPRDWRVENRHLRIYTMCKTCFSNS
IYYGDNTYHGHEDWLMYTDSKEFSNTYHNQGRLPDVDRHWSASCHHHHHHMGMSGS
18-06MDYKDDDDKKTNWQKRIYRVKPCVICKVAPRDWRVENRHLRIYNMCKTCFSNS
INNGDDTYHGHDDWLMYTDCKEFSNTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-07MDYKDDDDKKINWQKRTYRVRPCVICKVAPRDWRVVNRHLRIYNMCKTCFSNS
INYGDDTYYGHDDWLMYTDCKEYSNTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-08MDYKDDDDKKTNWQKRFYRVRPCVICKVAPRDWRVKNGHLRIYNMCKTCFSNS
IKYGDDTYYGHDDWLMYTDCKEFSNTYHNLGRLPNEDRHWSASCHHHHHHMGMSGS
18-09MDYKDDDDKKTNWQKRFYRVKPCVFCKVAPRDWRVENGHLRIYNMCKTCFSNS
LNNGDDTHYGHDDWLMYTDCKEFSNTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-10MDYKDDDDKKTNWQKRIYRVRPCVRCKVAPRDWRVENRHLRIYNMCKTCYSNS
INYGDDTYYGHEDWLLYTDCEEFSNTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-11MDYKDDDDKETSWHKLMYQVRPCVICKVAPRDWRVENRHLRIYTMCKTCFNNS
VNYGDDTHHGHNDWLMYADCNEFTNTCRNLARLPDEDRHWSASCHHHHHHMGMSGS
18-12MDYKDDDDKKTNRQKLIFRVKPCVICKVAPRDWQVENGHLRIYNMCKTCFINS
INNGDDTYHGHDDWLMHTDCTEFSNTYHNLGRLPGEDRHWSASCHHHHHHMGMSGS
18-13MNYKDDDNKKTNWQNRINRVRPCVICKVAPRDWCVKNGHLRIYNMCKSCFSDC
INYGDDTHYGHEDWLMYTDCKEFSNTYHNLGRIPEKDRHWSASCHHHHHHMGMSGS
18-14MATKDDDDKKTNRQKRIFRVKPCVICKVAPRDWRVRNGHLRIYNMCKTCFSNS
INYGDDTYYGHDDRLMYTDCMEFSNTYHNLGKLPDEDRHWSASCHHHHHHMGMSGS
18-15MDYKDDDDKKTNWLKRIYRVKPCVNCKVAPRDWRVKNRHLRIHNMCKTCYSNS
VNYGDDTYYGHDDWLMYTDCEEFSNTYPNLGSLPDEDRHWSASCHHHHHHMGMSGS
18-16MDYKDDDVKKTNWQKRIYRVRPCVICKVAPRDWRVENRHLRIYNMCKTCFSNS
INNGDDTYYGHDDWLMYTDSKEFSYTYHNLGWQPVEDRHWSASCHHHHHHMGMSGS
18-17MDYKDDDDKKTNWQKRTYRVRPCVICKVAPRDWRVKNRHLRIYNMCKTCFSNS
INYGEDTYYGHEDWLMYTDCEEFSKTYHNLGRLPGEDRHWSASCHHHHHHMGMSGS
18-18MDYKDDDDKKTNWQKRIYRVKPCVNCKVAPRDWRVKNRHLRIYNMCKTCYSNS
VNYGDDTYYGHDDWLMYTDCEEFSNSYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-19MDYKDDDDKKTNWLKRIYRVRPCVKCKVAPRNWKVKNKHLRIYNMCKTCFNNS
IDIGDDTYHGHDDWLMYADSKEISNTYHNLGRLPNEDKHWSASCHHHHHHMGMSGS
18-20MDYKDDDDKKTNWQKRIYRVGPCVICKVAPRDWRVENGHLRIYNMCKTCFGNS
INNGDDTNFGHDDWLMYTDCKEFSNTYHHLGGLPDEDRHWSASCHHHHHHMGMSGS
18-21MDYKDDDDKKTNWQKRIHRVGPCVICKVAPRDWRVRNRHLRIYNMCKTCFSNS
IKYGDDTYYGHDDWLMYTDCKEFSDTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-22MDYKDDDDKKTNRQKRIYRVRPCVICKVAPRDWRVENRHLRVYNMCKTCFSNS
IHYGDDTYHGHDDWLLHTDCKEFSNTYHQLGRMPDEARHWSASCHHHHHHMGMSGS
18-23MDYKDYDDKKTNWQKRICRVKPCVICKVAPRDWRVKNRHLRIYNMCKTCFSNS
IKYGDDTYYGHDDWLMNTDCKEFSNTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-24MDYKDDDDKKTNRQERLCRVRPCVFCKVAPRDWRVENKHLRIYNMCKTCFSNS
IKYGDDTYHGHDDWLMYTDCKEFSNTYHNLDRLPDEDRHWSASCHHHHHHMGMSGS
18-25MDYKDDDDKKTNWQKRIYRVRPCVICKVAPRDWRVKNRHLRIYNMCKSCFSNS
INNGDDTYYGHDDWLMYTNCEEFSSTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-26MDYKDDDDKKTNRQKRLFRVRPCVICKVAPRDWRVENGHLRIYNMCKTCFSNS
ISNGDDTYFGHEDRLIYSDCKEFSKTNHNPGRLPDVDKHWSASCHHHHHHMGMSGS
18-27MDYKDDDDKKTNWQKRNYWVRPCVICKEAPRDWRVENRHLRIYNMCKTCFSNS
IKSGDDTYYGHDDWLMYTDCKEFSDRYHNLARLPYEDRHWSASCHHHHHHMGMSGS
18-29MDYKDDDDKKTNWQKRNYRVRPCVICRVAPRDWRVKNGHLRIYNMCKTCFSNS
INYGDDTYYGHDDWLMYTDSEEFSNTYHNLDRLPDGDRHWSASCHHHHHHMGMSGS
18-30MDYKDDDDKKTNWQKPIYRVRPCVICKVAPRDWRVKNRNLRIYNMCKTCFSDS
IKYGDDTFHGHDDRLMFTDSKEFSNTYHDQGRQPDEDRHWSASCHHHHHHMGMSGS
18-31MDYKDDDDKKTNWQKRIYRVRPCVICKEAPRDWRVKNGHLRIYNMCKTCFSNS
INYGDDTYHGHDDWLIYKDCKEFSNMYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-32MDYKDDDDKKTNWQKRIYRVKPCVICKVAPRNWRVENRHLRIYNMCKTCYSNS
INYGDDTYHGHDDWLMYTGCKEFSNTYHNLGRLPDEVRHWSASCHHHHHHMGMSGS
18-33MNYKDDDDKKTNWQKHILRVRPCVVCKVAPRDWRVKNKHLRIYNMCKTCFSNS
INCGDDTHYGHKDWLIYTDCKESSKTYHDLGRLPDEDRHWSASCHHHHHHMGMSGS
18-34MDYKGDDDKKTNRQKRIYRARPCVICKVAPRDWRVEKRHLRIYNMCKTCYNNS
INYEDDTYHGHDDLGMYTDCKEFSNTYHDLGRLPDEDKHWSASCHHHHHHMGMSGS
18-37MDYKDDDDKKPNWLKRNHRVRPCMICKVAPRDWRVENGHLRIYTMCKTCFGNS
INYGDDTHHGHEDLWMNTDCKEYSYAYHNLGRLPHEDRHWSASCHHHHHHMGMSGS
18-38MDYKDDDDKKTNWKKRIYQVRPCVNCKVAPRDWRVENRHLRVYNMCRTCFSNS
INYGDDTFYGHDDWLLHTDCKQFSNTYHNLGRPPDEDRHWSASCHHHHHHMGMSGS
18-39MDYKDDDDKKTNWLKRIYRVRPCVVCKVAPRDWRLKNGHLRIYNMCKTCFSNS
TNNGDDTYYGHDDWLMNTDCKEFSNSYHNLGRLPDEDRAWSASCHHHHHHMGMSGS
18-41MDNKDDDDKKTNWQKCFYRVRPCVVCKAAPRDWRVENRRLRIYNMCKTCYSNS
INFGDDTHYGHDDWLMYSDSKEFSNTYHNLGRPPDEERHWSASCHHHHHHMGMSGS
18-44MDYKDDDDKKTNWQKRIYQMKPCVVCKVAPRDWRVKNRHLRIYNMCKTCFSNS
INYGDDSYYGHDDWLMNTNSKEFYNTYHNLGRLSDADRHWSASCHHHHHHMGMSGS
18-46MDYKDDDDKKTNRQKRIYRVRPCVICKVAPQDWRVENRRLRIYNMCKTCFSNS
INYGDDTHYGHVDWLMDMDSKEFSNTYHNLGRLPVEDRHWSASCHHHHHHMGMSGS
18-47MDYKDDDDKKTNWQKRIYRVRPCVVCKEAPRDWRMVDRHLRIYNMCKTCFSNS
NNYGDDTYYGHDDRLLYTDCKESSNTYHNPGGLPDEDRHWSASCHHHHHHMGMSGS
18-48MDYKDDDDKKTNWQKRIYRVRPCVICKVAPRDWRVENRHLRIYNMCKTCFSNS
IKYGDDTYHGHDDWLMNTDCKVFSNTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-51MDYKDDDDKKANWQKHIYRVRPCVICKVAPRDWWMENGHLRIYTMCKTCFSNS
INNGDDTYHGHEDWLMYKDCKEFSSTYHNLGRLPVEDRHWSASCHHHHHHMGMSGS
18-52MDYKDDDYKKTNWQKPIFRARPCVKCKVAPRDWWVENRHLRIYNMCKTCFNNS
INYGDDTYYGHDDWLVYTDCKEFSNTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-54MDYKDDDDKKTNWQKRIYRVRPCVICKVAPRDWRVENRHLRIYNMCKTCFSNS
INHGDDTYFGHDDWLMYTDRKEFSNTYYNLGRLPGEDRHWSASCHHHHHHMGMSGS
18-56MDYKDDDDKKTNWQKHIYRVRPCVRCKVAPRDWRVENGHLRIYNMCKTCFSNS
INYGDDTNYGHDDWPLYTDSKEFSNTYHNLDRPPDEDRHWSASCHHHHHHMGMSGS
18-57MDYKDDNDKMTNRQKRIYRVRPCVVCKVAPRDWRVENRHLRIYNMCKTCFSDS
IKYRDDTHHGHDDWLMYTDCMEFSNTYHNLGWLPDEDRHWSASCHHHHHHMGMSGS
18-60MDYKDDDDKKTNWLKRNYRVKPCVNCKVAPRDWRVKNRHLRIYNMCKTCYSNS
INYGDDTYYGHDDWLMYTDCEEFSNTYHNRGRLPDEDRHWSASCHHHHHHMGMSGS
18-61MDYKDDDDKKTNWRKRIYRVRPCVICKVAPRDWRVEDSHLRIYNMCKTCFSNS
INYGDDTYFGHEDWLMYTDCKEFSNTYHNLDRLPDENRHWSASCHHHHHHMGMSGS
18-62MDYKDDDDKKTNWQKRIYRVRPCVKCKVAPRDWRVENSHLRIYNMCKTCFSNS
INYGDDTHYGHVDWVMYTDCMKFSNTYHNLSRLPDENRHWSASCHHHHHHMGMSGS
18-63MDYKDDDDKKAYWQERIYRVKPCVICKVAPRDWRVKNRHLRIYNMCKSCFSNS
IIYGDDTYHGHDDWLMYTDCKEFFNTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-64MDYKDDDDKKTYWQKRIYRVKPCVICKEAPRDWRVKNRHLRIYNMCKTCFSNS
VNIGDDTYHGHDDWLDEYDCKEFSNTCHNLGRLPGEDKHWSASCHHHHHHMGMSGS
18-65MDYKDDDDKKTYWQKRIYRVRPCVVCKVAPRDWRVENRHLRIYNMCKTCFSNS
INYGDDTHYGHDDWLMNTDCKEFSNTYHNPGKLPDEDRHWSASCHHHHHHMGMSGS
18-66MDYKDDDDKKTNWQKSIYREKPCVVCKVAPRDWRVENGHLRIYNMCKTCYSNS
INYGDDTYYGHDDWLMYKDCKEFSNTYHYQGRLPEEDRHWSASCHHHHHHMGMSGS
18-67MDYKDDDDKKTNWQKRIYRVRPCVICKVAPRDWRVENRHLRIYNMCKTCFSNS
INNGDDTYHGHDDWLLYTDRKEFSNTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-68MDYKDDDDKKTNWQKRIYRVRPCVICKVAPRDWRVENRHLRIYNMCKTCFSNS
INYGDDTFYGHDDWLMYTDSKEFSNTYHNLGRLPDEDRHWSASCHHHHHHMGMSGS
18-72MDYKDDDDKKTIRQKRIYRVRPCVNCKVAPRDWRVENRHLRIYNMCKTCFSNS
INYRDDTYYGHDDWLIYTDCKEFSNTYHNLGRLPDKDRHWSASCHHHHHHMGMSGS
Fuller description of protocol for a round of selection:
In vitro selection and amplification
RNA was produced from the DNA library with T7 RNA polymerase and mRNA-displayed proteins were generated as previously described8,9. The linker, which connects the RNA 3’-terminus to the protein C-terminus, had the following sequence: (dA)21(triethylene glycol phosphate ester)3dAdCdCPuromycin (made using reagents from Glen Research, Sterling, VA). A 10 ml translation (400 nM template, Red Nova Rabbit Reticulocyte Lysate (Novagen, Madison, WI), according to the manufacturer’s instructions with 85 mM additional KCl, 0.85 mM additional Mg(OAc)2 and 25 nM 35S-methionine) incubated at 30°C for one hour yielded 7×1013 mRNA-displayed proteins after high-salt incubation (600 mM KCl, 25 mM MgCl2). The translation mixture was then diluted ten-fold into oligo(dT)cellulose binding buffer (1 M KCl, 100 mM Tris(hydroxymethyl) amino methane, 0.25% w/v Triton X-100, pH 8.0) and this mixture was incubated with 2 mg/ml oligo(dT)cellulose (Pharmacia, Piscataway, NJ) for fifteen minutes at 4°C with rotation. The oligo(dT)cellulose was washed on a chromatography column (Bio-Rad, Hercules, CA) with the same oligo(dT)cellulose binding buffer and then eluted with deionized water. The eluate was mixed with 2x Ni-NTA binding buffer (1x is 6 M guanidinium chloride, 0.5 M NaCl, 100 mM sodium phosphate, 10 mM Tris(hydroxymethyl)amino methane, 10 mM 2-mercaptoethanol, 0.25% w/v Triton X-100, pH 8.0) and then incubated with Ni-NTA agarose (Qiagen, Valencia, CA) for one hour at 4°C with rotation. The Ni-NTA agarose was then washed with Ni-NTA first wash buffer (8 M urea, 0.5 M NaCl, 100 mM sodium phosphate, 10 mM Tris(hydroxymethyl)amino methane, 10 mM 2-mercaptoethanol, 0.25% w/v Triton X-100, pH 6.3) and then with a gradient of increasing amounts of Ni-NTA second wash buffer (0.5 M NaCl, 10 mM Tris(hydroxymethyl)amino methane, 10 mM 2-mercaptoethanol, 0.25% w/v Triton X-100, pH 8.0), and then was eluted with Ni-NTA elution buffer (0.25 M imidazole, 0.5 M NaCl, 10 mM Tris(hydroxymethyl) amino methane, 10 mM 2-mercaptoethanol, 0.25% w/v Triton X-100, pH 8.0) for one hour at 4°C with rotation. EDTA was added to the eluate to give a concentration of 5 mM. The buffer was exchanged into Reverse Transcription buffer (50 mM Tris(hydroxymethyl) amino methane, 75 mM KCl, 3 mM MgCl2 pH 8.3) on a gel filtration column (Pharmacia, Piscataway, NJ). The mRNA-displayed proteins were then reverse transcribed with Superscript II (Gibco BRL, Rockville, MD) at 42°C for 30 minutes without a heat denaturation step. This sample was then exchanged into selection binding buffer (400 mM KCl, 20 mM HEPES, 4 mM MgCl2, 0.1mM EDTA, 2 mM glutathione, 1 mM glutathione disulfide, 0.25% w/v Triton X-100, pH 7.4) on a gel filtration column (Pharmacia, Piscataway, NJ), and then incubated with 100 µl of ATP-agarose (ATP attached via C8, 9 atom linker, cyanogen bromide activated cross-linked 4% beaded agarose (Sigma, St. Louis, MO)) at 4°C for one hour on a chromatography column (Bio-Rad, Hercules, CA). The column was then washed with 50 column volumes of selection binding buffer at 4°C for one hour and eluted with 12 column volumes of selection elution buffer (as selection binding buffer, but with an additional 5 mM ATP and 4.8 mM MgCl2, pH readjusted to 7.4) at 4°C for one hour. The eluted fraction was then brought to 0.1 M in NaOH, hydrolyzed at 90°C for 10 minutes, exchanged into deionized water on two successive gel filtration columns and amplified using PCR. Every round was assayed by SDS-PAGE to ensure that mRNA degradation had not occurred, and by scintillation counting of the 35S-methionine labelled proteins to measure the efficiencies of the various steps. These data were then used to determine the number of purified individual protein sequences introduced into the round one selection step as 6×1012 based on the proportion of total methionine incorporated into the mRNA-displayed proteins, and the efficiency of each of the subsequent purification steps.
This procedure was repeated for 18 rounds except that in rounds 10, 11 and 12 the PCR amplification was substituted by a mutagenic PCR amplification with an average mutagenic rate of 3.7% at the amino acid level. In rounds 14, 15 and 16 the amplification cycles were preceded by two ATP-agarose selection steps, and in rounds 17 and 18 the amplification cycles were preceded by three ATP-agarose selection steps. With successive selection steps the eluted fraction was exchanged into deionized water on a gel filtration column and purified on a denaturing Ni-NTA column and reverse transcribed as described above before being incubated with the ATP-agarose for the subsequent selection step. Also, the volume of the translation reaction was reduced to 1 ml except for round 1 in which it was 10 ml, and rounds 2, 10, 11 and 12 in which it was 5 ml. In the rounds of selection preceding the initial mutagenic amplification, incubation with a butyl agarose pre-column (Sigma, St. Louis, MO) was employed with the flowthrough being used for incubation with the ATP-affinity column.
K d by spin-filtration
Purified MBP-fusion proteins were exchanged into selection binding buffer by gel filtration and mixed with [α32P-ATP. These samples were incubated at 4°C for 30 minutes. 200 µl samples were then placed into Microcon-30 spin ultrafiltration devices (Millipore, Bedford, MA) and spun at 10,000g for 30 seconds with this filtrate being discarded, spinning at 10,000g for a further 45 seconds yielded a subsequent filtrate which, along with the unfiltered sample, were assayed by scintillation counting. This method was adapted from14. In competition experiments, the competitor was added after the incubation step, and then the solution was incubated for an extra 30 minutes at 4°C. Data were treated as above (Kd by Equilibrium Dialysis) and according to the method of Wang and von Hippel15 to measure the stoichiometry of the interaction.
K d by equilibrium dialysis
Purified MBP-fusion proteins were exchanged into selection binding buffer by gel filtration and 150 µl aliquots were placed on one side of a 14-16 kDa MWCO dialysis membrane in a Hoefer Scientific Instruments model EMD101B. Equal volumes of the same buffer containing diluted [α32P-ATP were placed on the other side of the membrane. After 24 hours at 4°C, samples were removed from each side of the membrane and assayed by scintillation counting. These data were fitted to y=c(x/(x+ Kd)), where x is the protein concentration, y the proportion of counts bound by the protein and c is the proportion of counts that are able to be bound by the protein, by an iterative algorithm (Deltagraph) to give the Kd. Competition experiments against unlabelled ATP or ATP analogues were performed similarly.
Back titration of hot ATP against cold ATP to give stoichiometry of interaction
This data was fitted to y = b+c((ANK+PK+1)
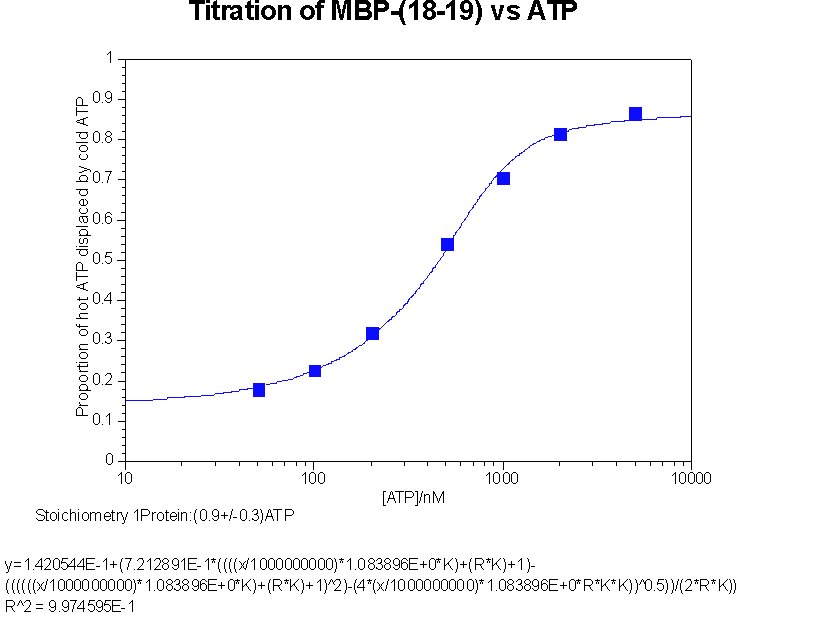
{kind=link}
- b is proportion of counts not passing through membrane, an iteratively fitted constant
c is proportion of counts bindable, an iteratively fitted constant
A is the ATP concentration
K is the association constant
P is the active protein concentration
N is the stoichiometry, an iteratively fitted constant
References :
- Roberts, R. W. & Szostak, J. W. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc. Natl Acad. Sci. USA 94, 12297– 12302 (1997).
- Sassanfar, M. & Szostak, J. W. An RNA motif that binds ATP. Nature 364, 550– 553 (1993).
- Wilson, D. S. & Szostak, J. W. In vitro selection of functional nucleic acids. Annu. Rev. Biochem. 68, 611– 647 (1999).
- Cho, G., Keefe, A. D., Liu, R. L., Wilson, D. S. & Szostak, J. W. Constructing high complexity synthetic libraries of long ORFs using in vitro selection. J. Mol. Biol. 297, 309– 319 (2000).
- Wilson, D. S., Keefe, A. D. & Szostak, J. W. In vitro selection of high affinity protein-binding peptides using mRNA display. Proc. Natl Acad. Sci. USA (in the press).
- Freedman, L. P. et al. The function and structure of the metal coordination sites within the glucocorticoid receptor DNA binding domain. Nature 334, 543– 546 (1988).
- Altschul, S. F. et al. Gapped BLAST and PSI-BLAST: a new generation of protein database search programs. Nucleic Acids Res. 25, 3389– 3402 (1997).
- Considine, D. M. (ed.) in Van Nostrand’s Scientific Encyclopedia 7th edn, 3067 (Van Nostrand Reinhold, New York, 1989).
- Liu, R., Barrick, J., Szostak, J. W. & Roberts, R. W. Optimized synthesis of RNA–protein fusions for in vitro protein selection. Methods Enzymol. 318, 268– 293 (2000).
- Keefe, A. D. in Current Protocols in Molecular Biology (eds Ausubel, F. M. et al.) Unit 24.5 (Wiley, New York, 2001).
- Cadwell, R. C. & Joyce, G. F. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 2, 28– 33 (1992).
- Wilson, D. S. & Keefe, A. D. in Current Protocols in Molecular Biology (eds Ausubel, F. M. et al.) Unit 8.3 (Wiley, New York, 2000).
- McCafferty, D. G., Lessard, I. A. D. & Walsh, C. T. Mutational analysis of potential zinc-binding residues in the active site of the enterococcal D-Ala-D-Ala dipeptidase VanX. Biochemistry 36, 10498– 10505 (1997).
- Jenison, R. D., Gill, S. C., Pardi, A. & Polisky, B. High resolution molecular discrimination by RNA. Science 263, 1425– 1429 (1994).
- Wang, Y. & von Hippel, P. H. Escherichia coli transcription termination factor Rho. J. Biol. Chem. 268, 13947– 13955 (1993).
Link collected : https://www.nature.com/articles/35070613